Hear about iJOBS from our students
About iJOBS
The key elements of the program are described below. You may also want to click here to view the presentation from the iJOBS informational forums.
The Rutgers University iJOBS Program exposes life science PhD students and postdocs to a range of non-academic and academic career options and empowers them to pursue their career goals. In 2014 Rutgers became 1 of only 17 schools in the country to be awarded an NIH BEST grant and iJOBS programming was later expanded to all campuses of Rutgers as well as Princeton, Rowan, NJ Institute of Technology, and Stevens Institute of Technology. iJOBS maintains research as the primary trainee focus while offering programming to broaden trainee's perspectives, experiences, and knowledge to facilitate the pursuit of academic and non-academic careers.
iJOBS Journey
Rutgers iJOBS participants follow a phased program to allow for maximum flexibility. Trainees can stay in Phase 1 as long as they want. If they would like to get more involved, trainees can choose to apply to Phase 2. Those that participate in Phase 2 for one year then progress through Phases 3 and 4.
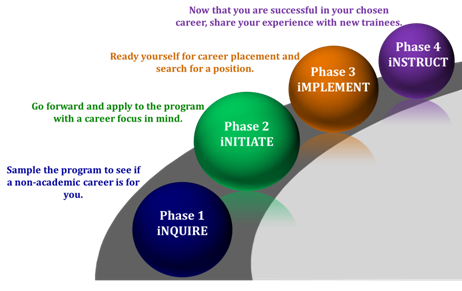
Phase 1 (iNQUIRE) programming is intended to lay the foundation for professional preparedness and provide insight into available non-academic career choices. Career panels, job simulations, company site visits, and networking events allow trainees to interact with professionals and be exposed to a number of different career tracts. Through the multi-day SciPhD workshop, participants will learn the business, management, and communication skills essential to career success.
Phase 1 program events are open to life science pre-doctoral students and post-doctoral fellows, although capacity is limited for some activities. Registration is required for each event through the EventBrite link that opens 4 weeks prior to the event at 12 pm (click here to see all upcoming and past events. Programming is very flexible such that most events are a stand-alone activity and a trainee can sign up for as many or as few events as they wish.
Phase 2 (iNITIATE) brings with it focus on a specific career path as well as more commitment to the program. Participants formally apply to the iJOBS program at this phase so that they can benefit from additional services. 1) Trainees accepted to Phase 2 are matched to a professional in their area of interest to shadow in an unpaid, part-time externship for up to 72 hours spread out over a semester. 2) Trainees are also allowed to register for a course in an area that will increase their skill set beyond science such as business, law, policy, regulatory affairs, etc… 3) Finally, Phase 2 trainees complete an Individual Development Plan and get ongoing feedback on their progress from a professional mentor that they are assigned.
Phase 2 applications require at least 12 hours of prior Phase 1 activities, a personal statement, permission from your PI, and a letter of recommendation from your PI or another faculty member with whom you have a long-standing relationship. Applications are open to graduate students and postdoctoral fellows. Graduate students must be in good academic standing (GPA > 3.0) and have completed the proposition qualifier. Postdocs can apply at any stage. Applications open each spring in April and programming begins the following September. Completion of Phase 1 and 2 components allows a trainee to obtain an Attestation of Completion Certificate from iJOBS.
Please note that iJOBS does not organize or promote full-time internships. Instead, we offer externships/shadowing in Phase 2. iJOBS administration strongly suggests that if a trainee does want an internship that they talk to their faculty PI and get their permission BEFORE applying to the internship. Furthermore, students should be close to completing their degree/thesis prior to applying for an internship.
Phase 3 (iMPLEMENT) is intended to serve as a bridge between career focus and career placement, with preparation for the job search and employment for trainees who have been through Phase 2. Individualized resume, LinkedIn and cover letter preparation, as well as interview practice and job searching tools, are provided.
Phase 4 (iNSTRUCT) is when iJOBS alumni are invited to give back to the program by becoming an industrial partner/mentor or friend to the program.
Program Tracks
To date, we have hosted programming focused on a number of career tracks and the list keeps growing in concert with trainee interest and increasing awareness of the vast options for PhDs in the workforce. Go to Past Events to learn more about each of these career paths.

Science & Health Policy
Intellectual Property
Research in Industry and Government
Clinical Trials & Regulatory Affairs
Contract Research Organizations
Health & Science Data Analysis
Finance and Equity Research
Entrepreneurial Science and Start-Ups
Medical Affairs
Science Writing and Communications
Science Education and Outreach
Non Profits
Consulting
Non-faculty Positions at Universities
Science Publishing
Venture Capital and Business Development
Science Journalism